フィル・マクヘイル、ティム・パロット、ジャン・クリストファーソン
小惑星に人工衛星を着陸させたり、火星でヘリコプターを操縦したりと、人類の創意工夫によって克服されてきた課題が次々と生み出されていますが、「宇宙は最後のフロンティア」というのは、スタートレックのファンなら誰でも知っていることです。
このチャレンジングなフロンティアの側面は、化学空間にも当てはまります。
実在する化学物質や仮想の化学物質のライブラリが増え続け、何十乗もの大きさになると、従来のケムインフォマティクスのツールやハードウェアのインフラでは対応しきれなくなります。
「この化合物はライブラリに含まれていますか」、「このコレクションの中で最も類似した10種類の化合物は何ですか」といった、一見単純でありながら根本的に重要なドラッグデザインの質問は、これらの超大規模空間において許容可能な応答時間で答えることが不可能ではありませんが、難しくなります。
本稿では、大規模な化学ライブラリを検索する際の現在の課題について考察し、次の最適で新規な合成可能な生物活性構造を追跡するために化学空間のはるか彼方を探索しようとしているドラッグデザイナー、メディシナルケミスト、ケムインフォマティシャンに、「火星のヘリコプター」レベルの画期的なソリューションを提供する可能性のあるアプローチについて検討します。
状況
製薬会社、公的なコンテンツ・リポジトリー、商業的な化学物質のサプライヤーやアグリゲーターが、既知の化合物、登録された化合物、公開された化合物、商業的に入手可能な化合物の様々なライブラリを統合して整理するにつれ、ファイル・サイズは数千万~数憶構造に近づくようになりました(例:eMolecules Plus 千万単位; PubChem, Sigma Aldrich 憶単位)。しかし、実在の化合物を合成可能な仮想化合物で補強することで、探索可能な化学空間の範囲と多様性を拡大したいという願望がありました。
Pfizer社は、社内のELN記録から得られた有効な反応変換情報と利用可能なビルディングブロックを組み合わせて、探索用の巨大な仮想ライブラリ(PGVL:1京の仮想、合成可能な化合物)を生成するコンビナトリアル技術を開発しました。他の企業やグループは、この反応ベースのコンビナトリアル・アプローチを基に、独自の知識やReaxysのようなソースからの文献ベースの反応情報を持ち込んで、ライブラリのサイズを拡大してきました。
DNAコード化ライブラリ(DEL)は、補完的なタイプの超大規模ライブラリを提供しています。バーチャルではなくリアルな化合物を、通常はスプリット&プールのコンビナトリアルケミストリーを用いて、数十億から数兆の化合物を含む大規模な混合物を生成し、各化合物の合成反応の実績をDNAタグにコード化します。DELは、ターゲットに結合する分子を濃縮し、結合しない分子を洗い流すシングルポットアフィニティアッセイでテストができます。結合した化合物は、PCRで増幅・精製することでデコンボリューション・デコードされ、続いてタグのDNA塩基配列を決定することで、個別の構造が特定されます。構造は実際の化合物のものだけですが、それを仮想ライブラリーで表現するのはも困難です。
2020年12月に開催されたNIH主催の超大規模化学データベースに関するワークショップでは、複数の参加者が、商業的(Enamine REAL Space 1010構造体など)、公的(BioSolveIT KnowledgeSpace 10の15乗構造など)、専有的(GSK GSKchemspace 10の26乗構など)な超大規模化学コレクションと、実在および/または仮想の合成可能な化合物を探索するためのライブラリが継続的に成長していることを強調しました。このような規模のライブラリを効率的に表現、保存、検索する方法への要求は、並行して高まっています。
課題
超大規模ライブラリの化合物を表現する最適な方法を見つけることは困難です。現在のケムインフォマティクスツールとコンピュータハードウェアでは、完全に列挙され、明示的に記述された超大規模な化合物セットを効率的に検索することはまだできず、1憶構造以上になると許容できないほど遅くなります。例えば、適切なメモリと特殊なハードウェアを使用すれば、10の6乗サイズの列挙型ライブラリは3.8MBを占有し、1秒という許容出来る時間で網羅的に検索することができますが、10の12乗サイズの列挙型ライブラリは3.8TBを占有し、検索には12日かかります。
このようなハードウェア/パフォーマンスの問題を解決するために、現在利用可能な技術は、フィーチャーツリーなどの縮小された化合物記述子を用いて、列挙されていないライブラリを表現・保存し、列挙されていないライブラリに対して最初に「ファジー・ファーマコフォア」類似性検索を実行することです。この方法では、より小さく、より焦点が絞られた、扱いやすいヒットセットを許容範囲内の時間で生成することができ、これらを列挙して、より詳細な構造および物理化学的特性の検索を行うことができます。
この手法は、リードの最適化やスカフォールドホッピングによく用いられるファーマコフォアの類似性/非類似性検索には適していますが、ライブラリ全体の完全な部分構造検索や、LogPやH-bond donorsの数などの重要な物理化学的またはトポロジー的特性を組み合わせた検索には対応していません。フィーチャーツリーは、環の置換パターンや立体化学を扱うことができないため、"この正確な構造は、列挙されていないセットに含まれているか?"や "データベースの中で、この潜在的なリード化合物に最も類似した化合物のセットは何か?"といった重要な質問に答えることができません。部分構造検索を提供する新しいアプローチもありますが、10の9乗以上のサイズのライブラリにどれだけ対応できるかについては疑問があります。
超大規模ライブラリのもう一つの課題は、可能な限りシームレスに共有する方法です。組織が完全な仮想ライブラリを受信して自社のサーバーやプライベートクラウドにロードし、さらに社内で処理や分析を行いたい場合、それぞれに複数のSMILESを含む大規模なファイルセットを送信することは、実行可能なソリューションではありません。
課題への対応
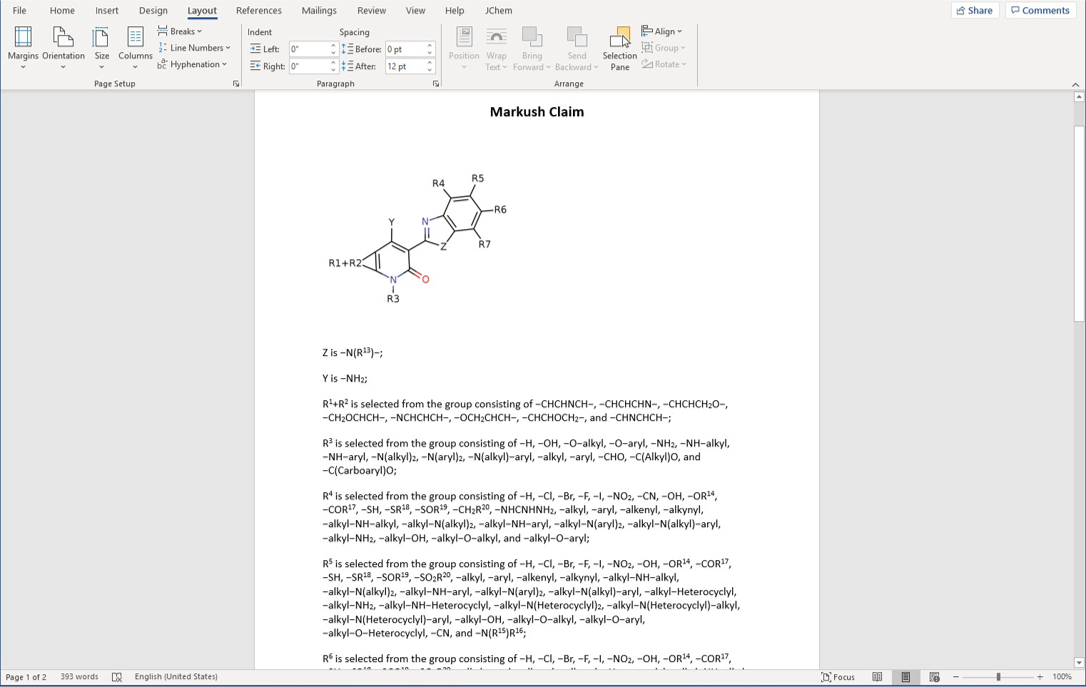
現在の離散的な列挙型構造を表現する方法は、超大規模なライブラリで使用するにはスペースを取りすぎる傾向があるため、すべての原子タイプ、連結性、立体化学を含む各化合物の構造を完全に忠実に再現する、よりコンパクトな非列挙型の表現が必要です。このようなニーズに応えるために、Compact Virtual Library (Compact VL)フォーマットが開発されました。
Compact VLは、MDL v2000のSDファイルをベースに、SDファイルのデータフィールド部に制限則や記述子を追加することで、反応変換情報をよりコンパクトに格納することができます。これにより、1つのSDファイルに完全なバーチャルライブラリーを格納することができます。例えば、反応物AとBがそれぞれ5Kずつの単純な二成分反応A + B → Cの場合、Compact VLでは25Mではなく10,001エントリーのSDファイルに結果のコンビナトリアルライブラリーを保存することができます。また、フィールドを追加することで、大きなコレクションの中のサブライブラリーを検索することができます。
このフォーマットのバーチャルライブラリは、現在入手可能なケムインフォマティクツールキットを用いて、反応変換ファイルとSDファイル内の試薬を組み合わせて生成することができ、一例として、Compact VLを生成するための KNIME ワークフローが作成されています。これにより、単一の SD ファイルとして共有できる仮想ライブラリが作成され、さらに分析および検索するために読み込むことができます。
現在、Compact VLフォーマットのライブラリを効率的に検索するための研究が進められており、類似性検索の強化や、完全な部分構造検索の追加が検討されています。大規模なライブラリをスケーラブルな検索システムに分散させることで、適切な検索性能が得られる可能性があります。
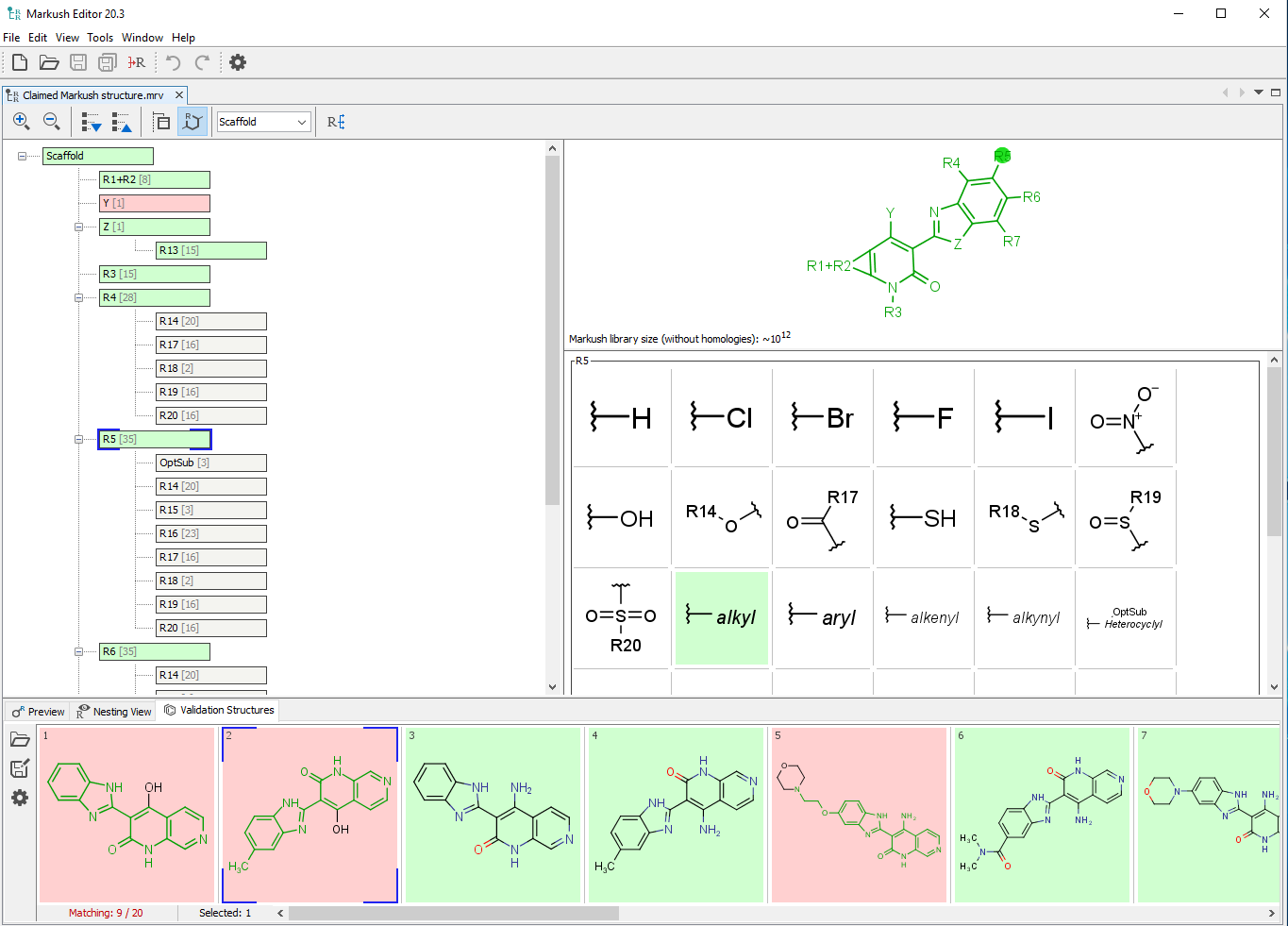
列挙されていないライブラリを検索するための新しいアプローチには、ファイザーが開発したLEAP2に似たアルゴリズムや、部分構造やMarkushベースの技術が含まれており、研究者がこれらの超大規模な仮想ライブラリを探索する際に、これまで答えられなかった重要な構造ベースの質問に対する答えを得ることができるようになるかもしれません。
※本稿はChemAxon公式ウェブサイトに掲載された下記の記事の日本語訳です。
Challenges in Searching Ultra-large Chemical Libraries by Phil McHale, Tim Parrott, Jan Christopherson
Compact VLと関連するKNIMEワークフローの詳細、およびこれらの新しい検索方法について、2021年6月9日よりウェブセミナーが開催されました。動画をご希望の方は以下よりお申し付けください。