
計算化学/創薬/合成容易性/分子設計
計算機で100万個もの魅力的な新規分子を一週間かけて生成したとします。そして翌朝、研究者の前には避けて通れない問いが残ります。
「このうち、実際に合成できる分子はいくつあるのか?」
その問いに、すぐには答えたくなくなるかもしれません。
計算機は分子構造を高速に生成できます。しかし、実験室のフラスコや試薬は、紙の上の分子設計や計算機の予測どおりには動いてくれません。合成容易性は、多くの創薬プロジェクトにおいて、分子設計から実験検証へ進む際の大きなボトルネックになっています。
この課題に対応するため、Synthetic Accessibility Score(SA Score)は重要な評価指標として広く用いられるようになりました。本稿では、主要な8つの合成容易性予測モデルについて、その技術的原理、強み、限界、そして適した利用場面を整理します。
合成容易性予測における3つの技術アプローチ
現在、合成容易性予測の分野では、大きく分けて3つの技術的アプローチが用いられています。
1. ヒューリスティック/ルールベース手法
専門家の化学知識と、手動で定義されたスコアリング関数に基づいて、合成の難易度を推定する手法です。
2. 機械学習/データ駆動型手法
大規模な化学反応データセットを用いて、統計モデルや深層学習モデルに合成パターンを自動的に学習させる手法です。
3. 逆合成ベース手法
逆合成解析を用いて、実際の合成経路を直接シミュレーションし、対象分子が実際に合成可能かどうかを判定する手法です。
SAScore:「見慣れた構造かどうか」に基づくスコアリング
SAScoreは、2009年にNovartis Institutes for BioMedical Researchの研究チームによって開発された指標です。SAScoreは、主に2つの観点から合成容易性を評価します。
分子の複雑性
分子量、環構造の数と種類、立体中心、その他のトポロジー特徴量など、構造的な複雑さを反映する特徴に基づいて計算されます。
フラグメントの見慣れ度
PubChem由来の100万分子を標準的なルールに従って部分構造フラグメントに分解し、大規模な化学データベース中で各フラグメントがどの程度頻繁に出現するかを記録します。一般的なフラグメントは加点され、希少なフラグメントは減点されます。
最終的なSAScoreは1から10のスケールに変換されます。1は非常に合成しやすい分子、10は非常に合成が難しい分子を意味します。このため、複数の分子間で合成容易性を直接比較することができます。
強み
1分子あたりミリ秒単位で計算できる高速性があります。また、ロジックが直感的であり、メディシナルケミストにとって理解しやすく、信頼しやすい点も利点です。大規模な計算インフラを必要とせずに導入できる点も実用的です。
限界
「構造的に複雑であること」と「合成が難しいこと」を明確に区別できません。複雑な構造を持っていても、確立された合成経路が存在する分子は、過度に難しいと評価される可能性があります。また、スコアは静的な構造特徴に基づいており、実際の反応可能性までは考慮していません。
BR-SAScore:SAScoreの弱点を補う拡張モデル
BR-SAScoreは、SAScoreの基本的な枠組みを維持しつつ、その主要な弱点である「実際の化学反応可能性を考慮していない」という点を補うために設計されたモデルです。
中核となる改良点は、フラグメントの由来に基づくスコアリング機構です。BR-SAScoreでは、「市販ビルディングブロックライブラリに由来するフラグメント」と「化学反応によって生成されるフラグメント」を区別します。さらに、逆合成ソフトウェアから得られる反応ルールとビルディングブロックに関する知識を、計算可能な化学フィンガープリントスコアとして符号化します。
この仕組みにより、BR-SAScoreは軽量なCASP代替手段として機能します。単なる静的構造特徴ではなく、経験豊富な合成化学者の直感により近いスコアを与えることを目指しています。
強み
実際の反応可能性をスコア関数に取り込むことで、SAScoreの本質的な盲点に直接対応しています。SAScoreの高速性と解釈性を保ちながら、精度を有意に改善しています。また、市販ビルディングブロックの利用可能性がスコアに直接反映されるため、実際の合成計画においてより実用的な判断材料になります。
限界
基本的には依然としてヒューリスティックモデルであり、実際の反応経路そのものをシミュレーションするわけではありません。性能は、使用する市販ビルディングブロックライブラリの網羅性と更新状況に依存します。また、反応ルールの範囲外にある非常に新規性の高いスキャフォールドに対しては、元のSAScoreに対する改善効果が限定的になる可能性があります。
SYBA:ベイズ確率に基づく合成容易性評価
SYBA(SYnthetic Bayesian Accessibility)は、ベイズ確率論に基づく合成容易性予測モデルです。このモデルが問うのは非常に単純です。
「ある分子の局所フラグメントは、合成しやすい分子に多く現れるのか、それとも合成しにくい分子に多く現れるのか?」
SYBAは、ECFP4分子フィンガープリントのフラグメントが、合成容易な分子群と合成困難な分子群のそれぞれにどの程度頻繁に出現するかを計算します。そして、その頻度差に基づいて合成容易性を判定します。各フラグメントには「合成しやすさへの寄与」を表すスコアが割り当てられ、それらを合計することで最終的な予測値が得られます。
強み
解釈性が高い点が大きな特徴です。各フラグメントがスコアにどのように寄与しているかを確認できるため、どの部分構造が合成上の難しさにつながっているのかを特定できます。
限界
各フラグメントが互いに独立であると仮定しているため、フラグメント間の結合形成そのものの難しさは考慮されません。また、合成困難な学習サンプルはアルゴリズムによって生成されるため、そのサンプルの品質がモデル精度に直接影響します。
SCScore:反応データから学習する合成複雑性
SCScore(Synthetic Complexity Score)は、この分野における一つの転換点といえます。専門家が定義したルールに依存するのではなく、大規模な実反応データから合成複雑性を学習するモデルです。
SCScoreは、次の化学的な前提に基づいています。
「生成物の複雑性は、反応物の複雑性以上でなければならない」
この「方向性制約」を用いることで、どのような構造が得やすく、どのような構造が作りにくいのかを学習します。
学習時には、ニューラルネットワークが反応ペア、すなわち「反応物 → 生成物」にスコアを割り当てます。このとき、生成物のスコアは常に反応物のスコアに一定のマージンを加えた値以上になるように学習されます。つまり、SCScoreは単なる「複雑性指標」を学習しているのではありません。合成とは構造的複雑性が増していく一方向のプロセスである、という化学的な常識を学習しているのです。
そのため、SCScoreのスコアは、静的な構造観察だけでなく、実際の合成ロジックに自然に整合したものになります。
強み
専門家定義のルールではなく、実際の反応データに基づいているため、スコアが実際の合成ロジックに沿いやすい点が特徴です。方向性制約により、恣意的な複雑性指標ではなく、化学的に意味のある学習目標が与えられています。また、多様な化学空間に対して良好な汎化性能を示します。
限界
明示的な合成経路は生成しません。分子がどの程度作りにくいかは示しますが、どのように作るかまでは提示しません。また、過去の反応データを用いて学習しているため、スコアは「過去に実施されたこと」を反映します。その結果、実際には合成可能であるものの、非常に新規性の高い化合物の合成容易性を過小評価する可能性があります。さらに、フラグメントベースの手法に比べると解釈性は低く、高い複雑性スコアを引き起こしている構造的要因を特定しにくいという課題があります。
RAscore:完全な逆合成計画に代わる高速な判定手法
仮想スクリーニングで数百万分子に対して完全な逆合成解析を実行することは、計算コストの面で現実的ではありません。
RAscore(Retrosynthetic Accessibility Score)は、この課題に対応するため、効率性を重視して開発されたモデルです。RAscoreは二値分類モデルであり、専門的な逆合成計画ツールであるAiZynthFinderによって、対象分子に完全な逆合成経路を計画できるかどうかを高速に判定することを目的としています。
強み
完全な逆合成解析よりもはるかに高速です。高スループットスクリーニングとの相性が良く、逆合成エンジンを自前で構築しなくても比較的容易に導入できます。
限界
出力は「可能/不可能」の二値判定に限られ、連続的な難易度スコアは得られません。また、モデルの性能上限は、基盤となるCASPツールの能力に完全に依存します。
DeepSA:分子を自然言語のように読むモデル
DeepSAは、自然言語処理(NLP)の技術を化学に応用したモデルです。分子のSMILES文字列を文章のように扱い、事前学習済み言語モデルを適用することで、「合成しやすい」または「合成しにくい」というラベルと、信頼度スコアを出力します。
重要なのは、DeepSAの学習ラベルが、Retro*などのツールによって計算された逆合成ステップ数に基づいている点です。10ステップ未満で合成可能と判断された分子は「合成しやすい」、10ステップを超える分子または計画に失敗した分子は「合成しにくい」とラベル付けされます。つまり、DeepSAは実験データそのものから学習しているわけではありません。
そのため、DeepSAが学習しているのは「アルゴリズムが経路を計画できるか」であり、「化学者が実験室で実際に合成できるか」ではありません。この違いは、実務上きわめて重要です。
強み
事前学習済みの分子言語モデルを活用することで、フィンガープリントベースの手法では捉えにくい複雑な配列パターンを学習できます。モデル読み込み後の推論は高速であり、大規模な仮想ライブラリにもスケールしやすい点が利点です。また、二値ラベルに加えて信頼度スコアも出力するため、モデルの確信度をある程度把握できます。
限界
ラベルが実験データではなく、アルゴリズムによる逆合成ステップ数から作られているため、モデルが学習しているのはソフトウェアが計画できるかどうかであり、化学者が実験室で実行できるかどうかではありません。また、二値出力であるため、同一クラス内で分子を順位付けしたり優先順位を付けたりするための粒度が不足します。学習分布から大きく外れた化学空間、特に新規性の高いスキャフォールドでは、性能が低下する可能性があります。
GASA:グラフニューラルネットワークによる重要部分構造抽出
GASA(Graph Attention-based assessment of Synthetic Accessibility)は、分子を原子と結合からなるグラフとして表現します。原子をノード、化学結合をエッジとして扱い、マルチヘッド・グラフアテンション層を用いて、合成容易性に関わる重要な構造特徴を自動的に学習します。さらに、結合レベルの情報を取り込むことで、分子全体の表現を強化します。
強み
高い汎化性能を持ちます。また、わずかな構造差が合成難易度に大きな違いをもたらす場合にも敏感に反応できます。解釈性にも優れており、原子レベルの寄与を可視化することで、どの原子や部分構造が合成の難しさに寄与しているのかを化学者が確認できます。
限界
学習ラベルはRetro*のような逆合成ツールに依存しており、実験結果との間には一定のギャップが生じる可能性があります。また、グラフアテンションの計算は従来のフィンガープリントモデルより重く、超大規模スクリーニングには必ずしも最適ではありません。
ChemAIRS SA Score Super Fast Mode:実際の合成ステップ数に基づく高速評価
ChemAIRS SA Scoreは、従来の複雑性ベースの指標を超え、実際の合成ステップ数、ビルディングブロックのコスト、反応可能性を取り込むことで、経験豊富な化学者が実際に合成経路を検討する際の判断に近い評価を提供することを目指しています。その深い評価能力の一方で、従来は計算速度とのトレードオフが存在していました。
この課題を解消するのが、ChemAIRSの新しいSA Score Super Fast Modeです。計算エンジンを再設計することで、速度を一桁向上させながら、ChemAIRSの中核的な強みである「分子複雑性の代理指標ではなく、実際の合成に関連するデータに基づく合成容易性スコア」を維持しています。
ChemAIRS SA Score Super Fast Modeの特徴
実世界データに基づく完全なデータ駆動型モデル
150万件以上のドラッグライク分子について、実際の最短合成ステップ数を用いて直接学習・検証されています。手作業で設計されたルールや、逆合成ソフトウェアからの間接的な予測ではなく、実験データに基づくため、実験室で起こる現実により近い予測が可能になります。
厳密なタイムスプリット検証
2023年以前のデータを学習・検証に用い、2023年以降のデータをテスト専用に保持しています。これによりデータリークを防ぎ、真に新規なドラッグライク分子に対する汎化性能をより正直に評価できます。
エンドツーエンドの非線形学習
ニューラルネットワークにより、分子構造と合成ステップ数の間に存在する複雑で非線形な関係を捉えます。従来モデルとは異なり、ChemAIRS SA Score Super Fast Modeは二値分類ではなく、実際の合成ステップ数と高い相関を持つ連続スコアを出力します。これにより、メディシナルケミストに対してより豊富な意思決定支援情報を提供できます。
数千万件規模の既知情報の統合
既知分子や市販出発物質に関する数千万件規模の知識を取り込むことで、他のモデルでしばしば見られる失敗を回避しやすくしています。たとえば、構造的には複雑に見えるものの、実際には確立された合成経路が存在する分子を過度に難しいと判断してしまう問題を軽減します。
モデル間の直接比較
2023年以降の独立テストセットを用いた体系的評価では、ChemAIRS SA Score Super Fast Modeが、予測スコアと実際の合成ステップ数との間で最も高いPearson相関を示しました。これは、同モデルが優れた予測精度を持つことを示しています。SCScoreと従来型のSAScoreも、強力な第二群のモデルとして続きました。
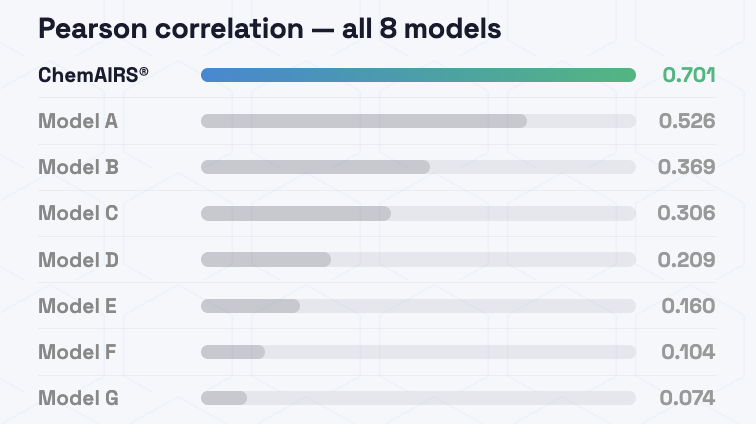
図1:モデル予測値と実際の合成ステップ数とのPearson相関
仮想スクリーニングにおける性能
より実応用に近い仮想スクリーニングシナリオ、すなわち詳細な追跡解析の対象として20,000分子を選抜する場面では、ChemAIRSは最も高いTop-20K選抜精度を達成しました。

さらに、ChemAIRSは高い安定性も示しました。サンプリングする分子数が増えても、精度が急激に崩れるのではなく、緩やかに低下します。実際の創薬プロジェクトにおいて、この安定性は非常に重要な特性です。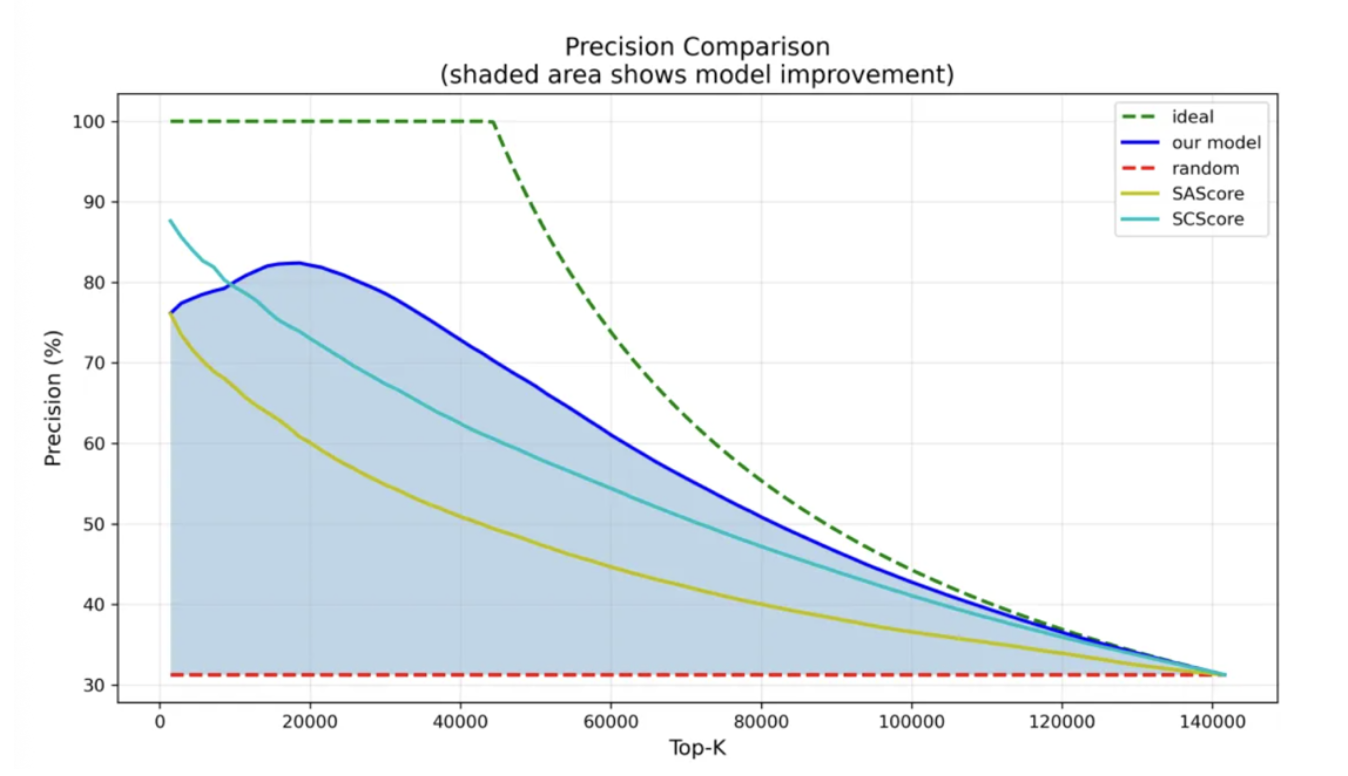
図2:テストセット上位10%分子における予測精度の変化
結論
SAScoreに代表されるルールベース手法から、SCScoreのようなデータ駆動型アプローチ、そして実際の合成ステップ数に基づくChemAIRSの新しい取り組みへと、合成容易性スコアリングはこの十数年で着実に進化してきました。
ChemAIRS SA Score Super Fast Modeは、約30分で100万分子を評価でき、合成可能性の高い候補分子を迅速に絞り込むことができます。ただし、Pearson相関が0.7であるということは、予測が完全ではないことも意味します。良好なスコアを示した分子であっても、実験室では依然として合成上の課題に直面する場合があります。
より高い精度と解釈性が求められる場面では、ChemAIRSの完全な逆合成推論モードの利用が推奨されます。これらのモードでは、より厳密な合成可能性評価、より高い説明性、そしてメディシナルケミストにとって具体的に利用可能な合成経路の提案が可能になります。
合成容易性評価の進歩は、AIが生成した分子と実験室での合成との間にある壁を少しずつ低くしています。そしてそれは、創薬をより速く、より賢く進める未来へとつながっています。

参考文献
- Ertl, P.; Schuffenhauer, A. Estimation of Synthetic Accessibility Score of Drug-like Molecules Based on Molecular Complexity and Fragment Contributions. J. Cheminform. 2009, 1, No. 8. doi:10.1186/1758-2946-1-8
- Chen, S.; Jung, Y. Estimating the Synthetic Accessibility of Molecules with Building Block and Reaction-Aware SAScore. J. Cheminform. 2024, 16, No. 83. doi:10.1186/s13321-024-00879-0
- Voršilák, M.; Kolář, M.; Čmelo, I.; Svozil, D. SYBA: Bayesian Estimation of Synthetic Accessibility of Organic Compounds. J. Cheminform. 2020, 12, No. 35. doi:10.1186/s13321-020-00439-2
- Coley, C. W.; Rogers, L.; Green, W. H.; Jensen, K. F. SCScore: Synthetic Complexity Learned from a Reaction Corpus. J. Chem. Inf. Model. 2018, 58, 252–261. doi:10.1021/acs.jcim.7b00622
- Thakkar, A.; Chadimová, V.; Bjerrum, E. J.; Engkvist, O.; Reymond, J.-L. Retrosynthetic Accessibility Score (RAscore) – Rapid Machine Learned Synthesizability Classification from AI Driven Synthetic Planning. Chem. Sci. 2021, 12, 3339–3349. doi:10.1039/D0SC05401A
- Wang, S.; Wang, L.; Li, F.; Bai, F. DeepSA: A Deep-learning Driven Predictor of Compound Synthesis Accessibility. J. Cheminform. 2023, 15, No. 103. doi:10.1186/s13321-023-00771-3
- Yu, J.; Wang, J.; Zhao, H.; Gao, J.; Kang, Y.; Cao, D.; Wang, Z.; Hou, T. Organic Compound Synthetic Accessibility Prediction Based on the Graph Attention Mechanism. J. Chem. Inf. Model. 2022, 62, 2973–2986. doi:10.1021/acs.jcim.2c00038
タグ: 合成容易性スコア/逆合成予測/創薬/プロセス化学/メディシナルケミストリー/計算化学
関連製品

関連モジュール




カタログ
「次世代AI化学合成設計プラットフォーム ChemAIRS」

関連ブログ








