Jan C. Christopherson, Chemaxon
数十億規模の化学ライブラリーの利用が標準化しており、クラウド技術やAI/MLの進展がその活用を加速させています。従来のツールでは対応が難しく、新しい表現形式や検索手法が求められています。今後の創薬には、これらの大規模ライブラリーを活用するためのインフラとツールの進化が不可欠です。
![Jan Christopherson[1]](https://patcore.com/hs-fs/hubfs/Jan%20Christopherson%5B1%5D.png?width=128&height=128&name=Jan%20Christopherson%5B1%5D.png)
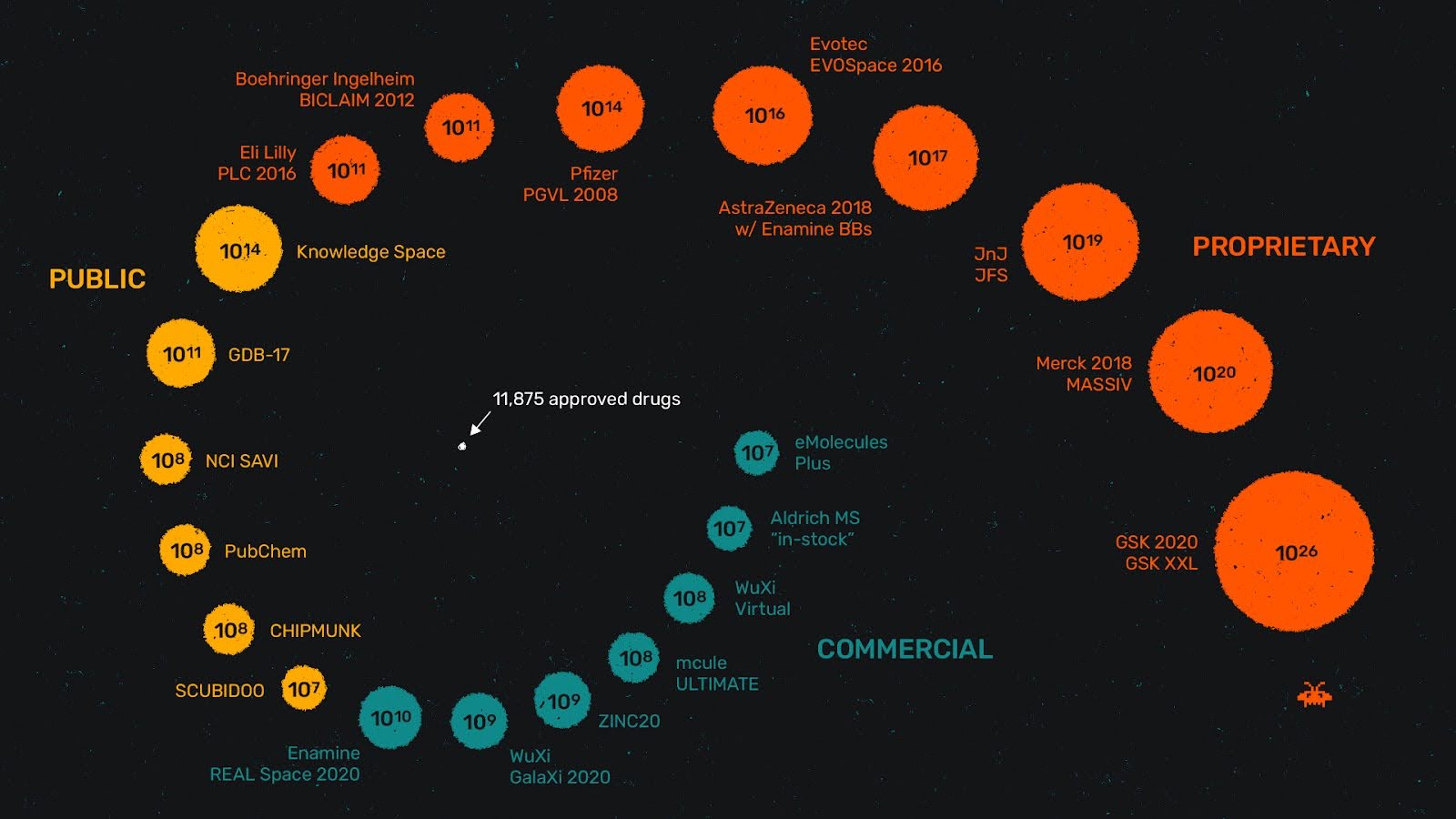
数十億規模のライブラリーが登場—これからはこれが常識!

図1. 利用可能な低分子のケミカルスペースは増え続けています。
近年、化学空間の探索は新たなスケールへと進化し、ライブラリーが数十億分子に達することも珍しくありません。この進化を加速させているのは、クラウドコンピューティングの利用、DELのような新しいスクリーニング手法、自動化されたラボなど、技術の進歩があらゆる面で進んでいることです。
また、「オンデマンド合成」ライブラリーの普及により、インシリコ法を超えてこれらの研究をさらに拡大することが可能になっています。AI/MLアプローチの普及に伴い、この規模のライブラリーが入力データ源としての役割を果たすことへの関心が高まり続けるのは必然と言えるでしょう。
ユニークな挑戦と魅力的な新しい表現の可能性
このような大規模なライブラリーは、情報学インフラに新たな課題をもたらします。多くのRDBMSカートリッジや類似のツールは、ユーザーの期待する速度とハードウェアの制限に対応しながら、この規模のデータを保存・検索することが困難です。
ライブラリーの表現には、データ量を圧縮するための非列挙形式が数多く開発されています。一般的には、ライブラリーを化学変化、特に反応と組み合わせた出発材料のセットとして表現します。これにより、少数の出発材料で、はるかに大規模なライブラリーを表現することが可能です。
この方法でライブラリーを保存するには、出発材料と、それらの組み合わせによる生成物の両方を検索するための新しい検索手法が必要です。代表的な手法にはLEAP2やFTreesがあり、これらについては以前に詳述しました。
このプロセスを実現するためには、まずライブラリーを作成し、それを適切に処理する必要があります。方法としては、1つ目に反応原子をラベルで置き換え、変換が起こる場所を示す方法があります。2つ目は、バニラ反応物を保存し、化学変換エンジンを使用して、必要に応じて構造ベースのマッチングと変換を実行する方法です。
ケミカル・スペース・ドッキングで生成物空間を効率的に調査
ドッキングは、多くの創薬チームにとって基本的なツールであり、読者にはその詳細な説明は不要でしょう。ドッキングは、ターゲットとする標的タンパク質に対する低分子の潜在的なポーズを決定し、ドッキングスコアを生成することで、複数の候補化合物の相対的な優先順位を決定する一般的なステップです。
FEPや他の量子力学的手法のような物理ベースのモデリングに比べると、ドッキングの計算コストは比較的少ないものの、新しい大規模ライブラリーにおいて実行するのは依然として困難です。
最近では、このような大規模ライブラリーに対する効率的なドッキング手法として「ケミカル・スペース・ドッキング」というアプローチが注目されています。この手法の詳細な紹介は本記事では省略しますが、詳細を知りたい読者は参考文献をご参照ください。
簡単に言うと、「ケミカル・スペース・ドッキング」は、フラグメントベースのドッキングを反復的に行う方法です。各ステップでは、前回の反復で得られた最良の結果が選択され、その反応性末端と適合する他のフラグメントが列挙されます。前の反復での最良のポーズは制約として使用され、次のドッキング反復でサンプリングするポーズの数を減らすことで、計算効率を向上させます。
プロセスの図解
-4.png?width=800&height=1338&name=Wiley%20Illustration%20(1)-4.png)
図1. ケミカルスペースドッキング
適切な表現を作成することは面倒な事前準備
このようなワークフローの導入は、間違いなく困難ですが、大変やりがいのある仕事です。可能な反応物のセットが与えられた場合、目的の変換が可能な反応物をどのようにして簡単に特定し、追跡できるでしょうか?
市販の多くのライブラリーがこのような形式で利用できるかもしれませんが、化学者は自分の組織独自の化合物を検索したいと考えることが多いでしょう。また、公開されている情報のみを使用していては、治療標的領域で競合他社に対する優位性を得るのは難しいでしょう。
そのため、化学の文脈において適切な検索と置換を行う能力が必要です。ChemaxonのReactorは、この目的のために特別に設計されたツールであり、ユーザーは望むマッチング条件と修正された出力表現を、業界最高水準の化学的精度で入力できます。これにより、化学ドッキングモデルワークフローのための適切な入力データを確実に生成できます。
ケーススタディ カルボン酸からのアミド合成
カルボン酸からアミドを合成する小規模なケーススタディを準備しました。
ChEMBLから最初の1000個の低分子をフィルタリングし、まず空の構造をすべて除去しました。
図2には、手順を追ったウォークスルーが示されています。プロセスの複数のステップを設定する際の追加の労力や、いくつかの測定可能な数値が際立っています。
特に、R-グループキャッピング法の最終ステップは、フィルター前の構造認識に基づく手法に比べて、実行速度が数桁遅いことが明らかになりました。
第二に、Rグループまたは類似のエンドキャップを使用した列挙は、はるかに多くの化合物を生成することになります。これは最初は魅力的に見えるかもしれませんが、実際には、ユーザーまたは別のフィルタリングプロセスのいずれかが後で除去しなければならない、はるかに不合理な化合物を単にリードすると考えています。
Synthon 表現の本質的な限界
反応部位を表現する方法の一つは、変換によって取り除かれる原子を取り除き、「R」基の表現に置き換えることです。これらは 「Synthons 」と呼ばれます。
これは特定の反応の反応性を追跡する簡単な方法ですが、多くの欠点があります:
- 反応物上に複数の一致する部位がある場合、どのように表現が扱われるべきかという疑問が生じます。
- 文脈から外れると、反応部位が何であるかについての重要な情報が削除されます。
- 反応性ルールの更新が行われた場合、バックトラックして一致する反応物のセットを再生成するのは難しいかもしれません。
- 複数の反応が範囲に含まれる場合、複数の異なる反応を受ける可能性のある化合物をどのように扱うかという問題が生じます。
- 同じ化合物に複数のR基が結合している場合、R基 ライブラリーの管理が必要になります。
- あるいは、同じ反応物を異なるR基で複数回保存すると、保存効率が低下し、注意深く管理しないと冗長なドッキング計算をリードする可能性があります。
単純なアプローチでは、一致する部位にRグループを適用することが一般的ですが、化学者として、それだけでは反応性が保証されないことを理解しています。構造全体や部分構造の特性を計算し、それをフィルタリングや優先順位付けの条件として活用できるアプリケーションを用いることで、反応性予測の精度を向上させることができます。
市販のライブラリーの多くは、長年にわたる知識の蓄積を通じて検証されており(多くの場合、ライブラリー全体の合成成功率とともに報告されています)、そのような徹底した検証には相応のリソースが必要です。しかし、独自のライブラリーを構築しようとする多くの組織には、そのリソースが不足しているのが現状です。
そのため、マッチングを行うソフトウェアツールが、分子の特性を候補基準として利用する単純なアプローチから、機械学習を取り入れた具体的な逆合成ツールに至るまで、成功の可能性を予測するうえでユーザーを支援することが重要です。
反応ベースのコンビナトリアル表現は、ケミカル・スペース・ドッキング以外にも有用
新しいアプローチが次々と開発される中、これまで述べてきたような表現の使用例も増加しています。現在、特に注目されているユースケースの一つがDNAでコードされたライブラリーです。このライブラリーは本質的に組み合わせ的な性質を持つため、これまで説明してきたライブラリーの表現を生成し、それに対する検索を成功させるためのツールは、DNAライブラリーにも関連性があります。
大規模なライブラリ生成への挑戦
Chemaxonでは、大規模な列挙の利点と課題についての調査を引き続き進めていくことに関心を持っています。組合せ列挙に必要な計算リソースは、一見すると困難に思えるかもしれませんが、AWS Spot Instancesのようなコスト効率の高いプラットフォームが利用できるため、克服可能な問題です。しかし、この取り組みは、適切なバッチ処理とワークフローの管理といった課題にもつながります。場合によっては、クラウドサービスのネイティブなタスク管理と配信サービス、例えばロードバランサーのような機能が好まれることもあります。その他のケースでは、JChem Microservices Reactorのタスクマネージャー機能のように、列挙サービスとより密接に統合されたツールがユーザーにとって有益です。
 |
ReactorReactorは大規模で利用可能なケミカルスペースを探索するための包括的なバーチャル反応エンジンです。一般化した反応式を用いてバーチャルライブラリーを作成するためのソフトウェアです。単にバーチャルな分子を作るのではなく、化学的に意味があり、合成できる可能性の高い化合物を生成するユニークなアプローチを採用しています。このアプリケーションは、デスクトップアプリケーション、コマンドラインツール、ワークフローツール(KNIME、Pipeline Pilot)、プログラムアクセス用のJavaまたはREST APIとして利用できます。 |

|
JChem extensions for KNIMEChemAxonツールのライセンスと、JChem Extensions for KNIME(※1)を用いることで、研究者がKNIMEワークフロー内でChemAxonツールを活用できるようになります。 ※1 販売元:Infocom http://infocom-science.jp/product/detail/jchemextensions.html |
 |
JChem Enginesケミカルインテリジェンスでオブジェクトリレーショナルデータベース管理システムを強化 JChemテクノロジーは、化学表示、検索、保存、構造および非構造データ管理機能のための高度な実装を提供します。データ管理機能は、ChemAxonアプリケーションの多種多様な統合システム全体に広く埋め込まれたコアエンジンであり、研究情報科学システムが依存する高速で目に見えないバックボーンを提供します。「主力」として、さまざまなデータベースエンジン(Oracle、PostgreSQL、MySQL、MSSQLなど)に格納されたデータを処理でき、複雑な検索テクノロジー、幅広いオプションスペース、および化学構造を処理するための高度な機能を提供します。 |