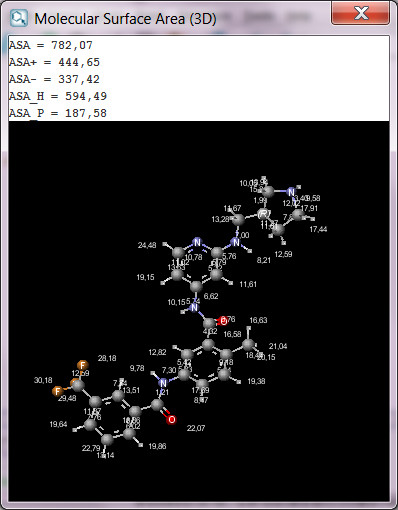
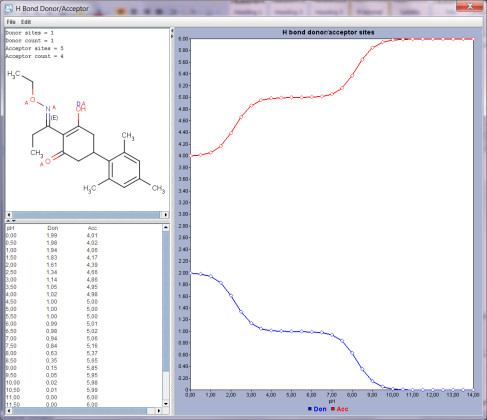
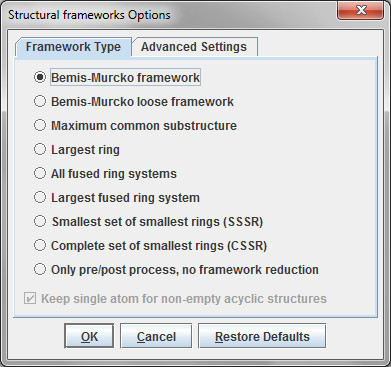
SARvision | Biologicsは既存のバイオロジックスソフトウエアの隙間を埋めるデスクトップアプリケーションで、ペプチドや核酸などバイオポリマーのデータを読み込み整理する為の基盤ツールと配列と活性の関連を見出す様々なツールを提供します。
配列解析に最適化されたスプレッドシートが提供され、非天然アミノ酸や化学的に修飾された残基を含む、RNA、タンパク、ペプチドといったバイオポリマーを取り扱うことが出来ます。
特徴
ペプチド、抗体、RNAに関する研究をを支援
非天然アミノ酸、環化、化学修飾を含むペプチドの取り扱いが可能
ヒートマップによる活性の視覚化機能
興味のある領域のハイライト
活性に大きな影響のある変異の探索(Logo Plot、Invariant Map、Mutation Cliff)
配列データの読み込みとアライメント(自動および手動)
ANARCIに基づいたナンバリングアルゴリズムを実装し抗体に自動的にナンバリング(Kabat、Chothia、Enhanced Chothia、IMGT、AHoに対応)
何百もの配列を検討可能
参考文献
オンラインチュートリアル
生物学的製剤のシーケンスとデータを保持するために今でもExcelを使用していますか?
こちらのビデオでは、SARvision | Biologicsを使ってより簡単に、ペプチド構造活性相関から多くの知見を抽出する方法をご紹介します。
クイックスタートガイド
残基と修飾子のテーブルの準備
これらの2つの重要なファイルの説明
残基/修飾子テーブル
データビュー
シーケンステーブルビューでシーケンスとデータを視覚化する方法。
シーケンステーブルの操作
Logo and Bar Plots
Logo Plotsは、ペプチドのSARの傾向や、抗体の表現型の特徴を分析するための優れたツールになります。
Logo/Bar Plotの操作
Invariant Maps
Invariant Mapsは、配列アラインメントの各位置でどの残基が試行されたか、およびその回数を示します。
Invariant Maps
Mutation cliffs
Mutation cliffsは、単一の位置でのみ異なる配列のペアを識別し、突然変異と配列の位置の両方でそれらを編成するために設定されます。
Mutation cliffs
サブセットグループの操作
あるビューを別のビューでフィルタリングする方法。
サブセットグループの操作
抗体のナンバリング
抗体設計におけるデータへの配列の関連付けを容易にするためのアプリケーションノート。
Antibody Design
資料ダウンロード
『機械学習による配列からのペプチド最適化』という資料(全編英語表記)をご用意しております。下記「DOWNLOAD」ボタンをクリックしてiPROSからダウンロードをお願いします。
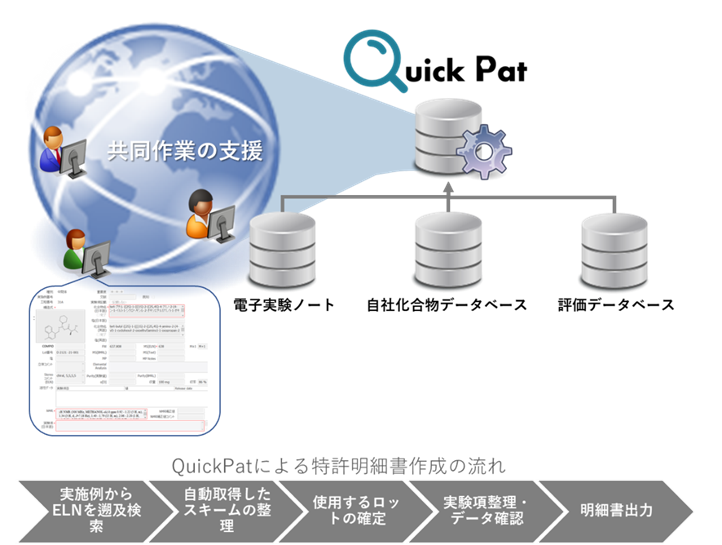
SARvision SMは、化学者にとって重要な仕事のひとつである構造活性相関の検討を支援するソフトウェアです。
SARvision SMの特長
構造式群から自動的に共通部分骨格を抽出し、骨格ごとに分類したツリーを提示
骨格を物性や活性データでソートしたり、スライダーを使った動的な絞込み
物性値を計算し、活性値と共に迅速にグラフ化(構造情報とリンク)
共通部分構造と置換基を整理しRグループテーブルを瞬時に作成
豊富なグラフィック機能(棒グラフ、2D散布図、3D散布図)
RグループテーブルをEXCELやWORDへ出力し効率的にレポートを作成(コマンドラインオプションをサポートしており、他のソフトウエアとの連携も可能)
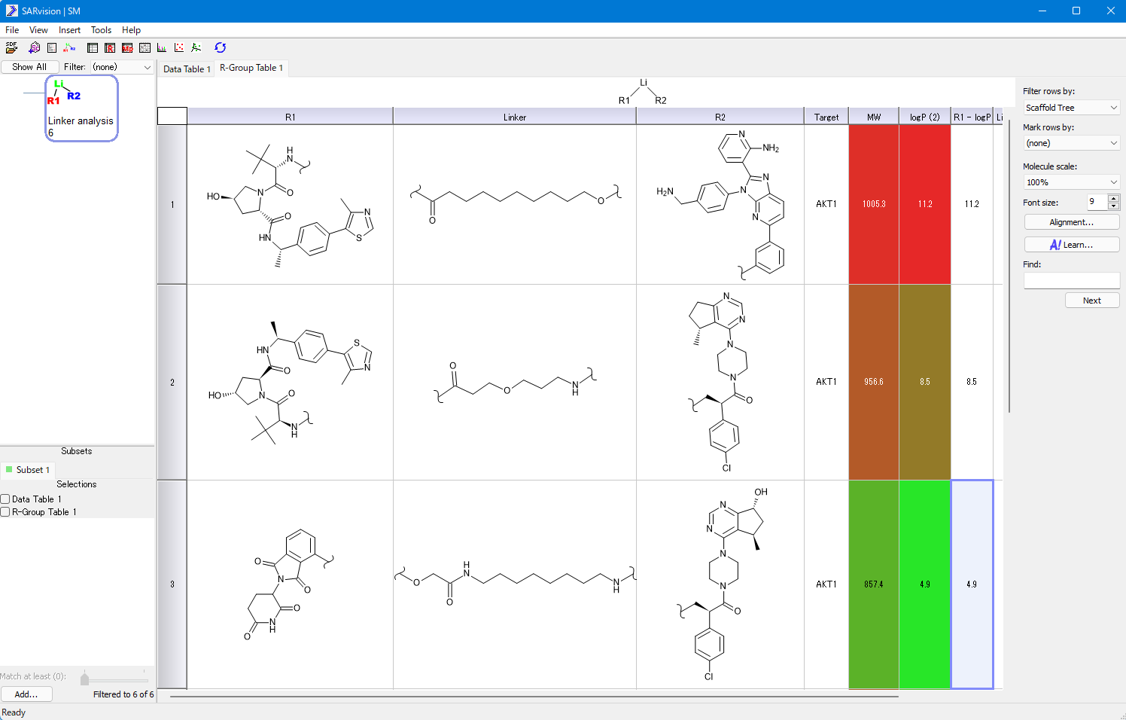
■NEW■ PROTAC解析モジュール新登場!
リンカー解析モジュールオプションが追加され、バイリガンドのSAR研究をルーチン化、かつ容易に実行できるようになりました。この新しい解析ツールはPROTACを研究するために設計されていますが、リンカーによって結合された2つのリガンドであれば、どのような分子にも対応可能です。
詳細につきましては、
「PROTAC解析モジュール」ページ をご覧ください。
*分子をリンカーと2つのリガンドに分解し、SARvision|SMのSAR Tableで簡単に解析できるようにします。PROTAC分子の場合、E3リガーゼモジュレーターが特定され、左側に配置されます。リンカーは、2つのリガンドの間に水平に伸ばされて表示されます。残りの部分、ターゲティング弾頭は右向きに表示されます。
チュートリアルビデオ
Quick Start - Part 1
構造データの読み込みと骨格ツリーの構築
VIDEO
Quick Start - Part 2
テーブルの作成とレポートの生成
VIDEO
Quick Start - Part 3
グラフの作成、データの視覚化
VIDEO
Rグループテーブルの作成
VIDEO
解説ビデオ 1
MMP(Matched Molecular Pair)解析機能
VIDEO
解説ビデオ 2
Scaffold Hopping機能
VIDEO
解説ビデオ 3
画像の構造を構造式に変換して取込み
VIDEO
解説ビデオ 4
CDD Vault連携機能
VIDEO
ほとんどの研究者にとってハイスループットスクリーニング(HTS)のデータ分析は今日もなお挑戦的なタスクとなっています。HTS実験に携わる研究者は、データの分析・視覚化・解釈の為に洗練された実用的なソフトウェアを求めています。これらの解決策を説明するために本レポートでは、ChemBankからのアッセイデータセットをどのようにして簡単にSARvisionSMに取込み、素早くSARテーブルを生成し、骨格を抽出し、動的なプロットが1つのデスクトップアプリケーションで行えるかを解説します。
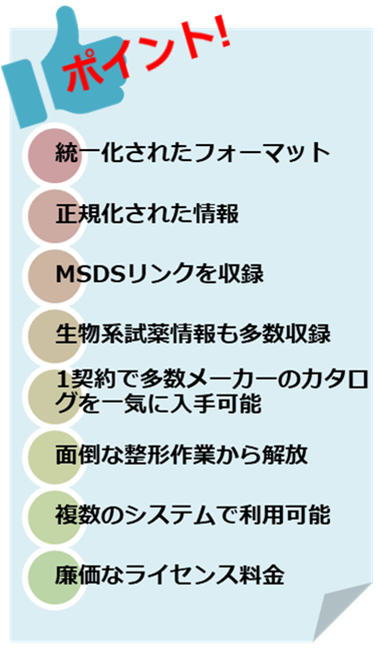
試薬カタログの課題を一気に解決!
製薬/化学会社では、購入する試薬の調査や危険物管理など、国内法令に準拠した適正な管理を行うために、個々の試薬メーカーよりカタログを取り寄せ、大変な手間をかけて試薬管理および購買システムで必要とされる情報の整備を行ってきました。しかし、海外製の市販試薬カタログデータは年間使用料が高額であり、また国内法令やSDS情報の記載が十分でないことがあります。こうした課題を解決するために、SMARTSは開発されました。
化学構造式が製品に紐づけられていますので、インシリコ手法で実験に有用な試薬を絞り込み、効率的に試薬を調達することが可能になります。
約3,200万件の商品と1,100万件の構造式を収録
収録ベンダー(50音順・法人格省略)
アブカム*
ATTO NEW!
関東化学
キアゲン*
キシダ化学
コスモバイオ*
シグマアルドリッチジャパン合同会社
重松貿易
タカラバイオ*
東京化成工業
ナカライテスク
ナミキ商事
フナコシ*
富士フイルム和光純薬
渡辺化学工業
※上記は販売元であり、製造メーカーは多数収録されています。
* 構造式データが提供されないベンダーです。可能な限りSMARTSの構造式と商品情報の紐づけを行っています。
主な収録情報
SDファイル(構造式)
SMARTS No.(SMARTS独自の化合物ID)
InChIKey
IUPAC名
化合物構造式(標準化済み構造式)
テキストファイル(商品情報)
SMARTS No.(SMARTS独自の化合物ID)
化合物構造式(標準化済み構造式)
販売元名称
販売元カタログ番号
製品名
製品別名
容量
規格
カタログ表示価格
法規制情報(毒劇法、薬機法、消防法など)
法令コード
法令正規化略称
化管法号番号
SDSリンク(URL)
保存条件
ライセンス形態
利用ユーザーに基づく年間ライセンス価格(サイトライセンス有り)
最高年4回のアップデート
契約に際して事前審査があります。(試薬ベンダー様等へは販売できない場合があります。)
ご注意
すべてのデータ項目に値が収録されているわけではありません。実際にデータが提供されるデータ項目はベンダー・製品毎に異なります。
法規制情報はベンダーが提供した情報を基に表現を統一して提供しています。その正確性、網羅性を保証するものではありません。
お客様個別のご依頼に基づき、CAS登録番号の調査・提供に対応致します。
資料ダウンロード
下記「DOWNLOAD」ボタンをクリックしてiPROSからダウンロードをお願いします。