Charge Plugin:パーシャルチャージ、分極率、電気陰性度の計
Charge plugin:パーシャルチャージの計算

Partial charge distributionはイオン化定数や反応性、ファーマコフォアパターンなど多くの物理化学的プロパティーを決定づけます。Charge pluginは改変されたGasteiger の方法に基づき個々の原子のパーシャルチャージを計算します。(J.Gasteiger and M.Marsili: Tetrahedron Vol. 36. , pp. 3219-3288 (1980), M.Marsili and J.Gasteiger: International Symposium on Aromaticity, Dubrovnik, Yugoslavia , Sept (1979), Croat.Chim.Acta. (1979) )トータルチャージはシグマおよびパイチャージコンポーネントから計算されます。結果をMarvinSpace上にマッピングすることもできます。
Polarizability plugin:分極率の計算
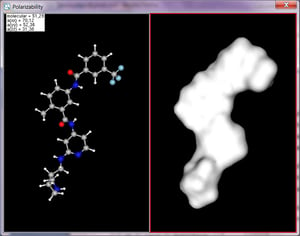
Polarizability pluginはパーシャルジャージの効果を考慮に入れて分極率を計算します。
Orbital electronegativity plugin:電気陰性度の計算
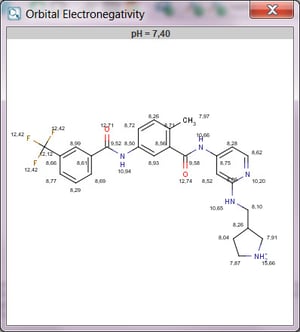
Orbital Electronegativity Pluginは原子のパーシャルチャージディストリビューションから電気陰性度を計算します。
Conformation Plugin:コンフォマーの発生、重ね合わせ、MD計算
Conformer plugin:コンフォマーの生成
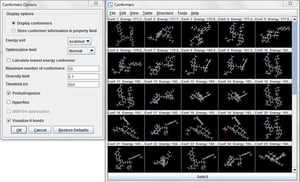
Conformer pluginは安定した3次元構造を発生します。コンフォマーの安定性は分子動力学力場のフレームワークにおいて計算されたエネルギーの比較により評価されます。3次元構造は分子の物理化学的性質や反応に大きな影響を与えます。また、Dreiding あるいはMerck Molecular force-fields (MMFF94) を用いて最少エネルギーのコンフォマーを得ることも可能です。
3D Alignment plugin:分子の重ね合わせ
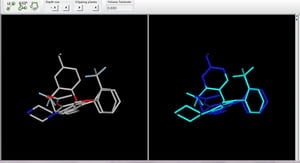
3D Alighnment Plugin 分子を重ね合わせする際、2種類の異なるアプローチを用いることができます。1つ目は入力されたコンフォメーションを用いるリジットアプローチで、2つ目は三次元座標をオンザフライで調整して重なりを最大化するフレキシブルアプローチです。
Molecular dynamics plugin:分子動力学計算(MD)
分子動力学シミュレーションは分子構造を動的に変化する実態として説明することを目的としています。柔軟な分子の構造はコンフォマーを用いるよりも、分子動力学シミュレーションによって求めた軌跡を用いる方がより正確に説明ができます。Molecular Dynamics Pluginは分子力学の力場を用いて原子核の位置を計算します。計算結果の軌道は分子の羅列やアニメーションにより確認することができます。
機能一覧
-
Display: 表示モード
-
Frames: 軌跡フレームの個別表示
-
Animation: アニメーションにより軌跡を表示
-
-
Forcefield: 計算に用いる力場
-
Integrator: ニュートン力学を解く為に用いる積分形式
-
Simulation steps: シミュレーションステップ数
-
Step time: シミュレーションステップ間の時間(fs)
-
Initial temperature:系の初期温度(K)
-
Start time of display: 表示される最初のシミュレーションフレームの時間(fs)
-
Frame interval: シミユレーションフレームの表示間隔(fs)
Huckel analysis Plugin:局在エネルギー等の計算
アロマティックセンターに対する電子吸引性および核吸引性の作用に関する局在エネルギーL(+)とL(-)はヒュッケル法によって計算されます。 小さな(+)とL(-)はアロマティックロケーションの反応性が高いことを示します。E(+) あるいはNu(-) における原子の順番はそれらの局在エネルギーに応じて調整されます。また、トータルΠエネルギー、Π電子密度、および総電子密度もヒュッケルの方法により計算されます。
Refractivity Plugin:モル屈折率の計算
Viswanadhanらが提案したアトミックメソッドに基づきモル屈折率を計算します。モル屈折率は分子の体積および薬物受容体との相互作用に重要な影響を与えるロンドン分散力と強い関連性があります。
Geometry Plugin:分子の様々な幾何学的パラメータを計算
Topology Analysis Plugin:トポロジー分析
Topology Analysis は分子の位相幾何学的な指標を計算します。例えばリングに関しては、リング数、脂肪族・芳香族リングの数、縮合環の数、最小・最大のリングなどの値を算出します。その他ローテータブルボンド数、ステレオセンター数や各種インデックス(Wiener, Szeged, Platt, Randicなど)を算出します。
幾何学的指標の計算
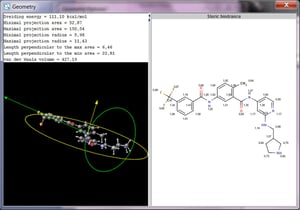
Geometryは分子の幾何学的な指標を計算します。計算に供する入力分子はユーザーが入力した分子とするかエネルギーが最小のコンフォマーとするかをユーザーが選択できます。
-
Dreiding energy:3次元構造(コンフォメーション)の安定性に関連するエネルギー
-
Hindrance:電子対共有範囲と幾何学的距離より計算した原子の立体障害
Polar Surface Area Plugin:極性表面積の計算
極性表面積(2D PSA)は分子の極性原子によって形成されます。 PSAは膜を介する分子の受動輸送と高い相関関係を示し、医薬分子の輸送量に関する予測に利用できるディスクリプターです。計算結果は実用上3D PSAとほぼ同じ結果を得られますが、3D PSAと較べて約100倍高速に計算できますので、大規模なバーチャルライブラリのバイオアベイラビリティースクリーニングに適しています。
Molecular Surface Area Plugin:分子表面積の計算
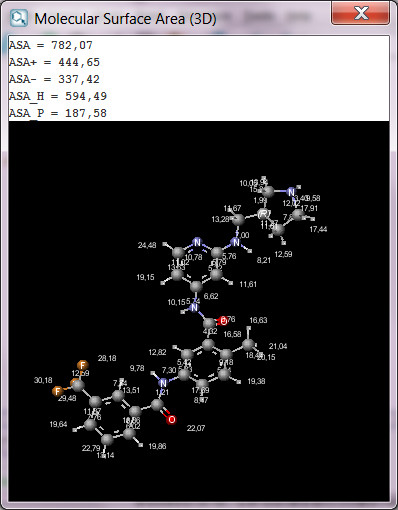
Molecular Surface Area は Van der Waals と solvent accessibleの2タイプの分子表面積を計算します。
Hydrogen Bond Donor-Acceptor (HBDA) Plugin
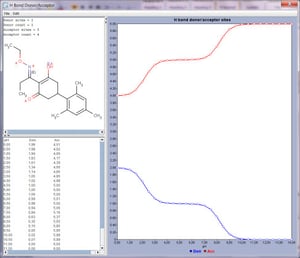
入力構造の個々の原子の情報、および分子全体のドナー、アクセプターの数を表示できます。またpH毎のカウント値をグラフ表示できます。
Structural framework Plugin:骨格の抽出(MCS,Bemis-Murcko framework)
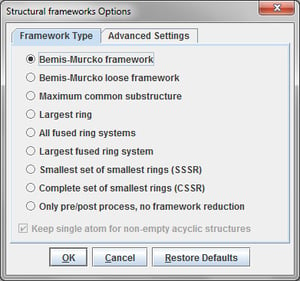
構造の比較において、骨格の抽出は、構造検索やクラスタリングと並んで重要な役割を担っています。良く知られた方法としては、構造ベースのクラスタリングを可能にするBemis-Murcko frameworkがあげられます。本ツールでは様々なパターンのフレームワークを抽出できます。
-
fused ring systems
-
largest fused ring systems
-
the complete set of smallest rings in a structure
また、本ツールは複数の分子から最大共通部分構造(MCS:maximum common substructures)を計算できます。
関連リンク(英文)
Documentation
API Docs
Training & Examples
Support & FAQ